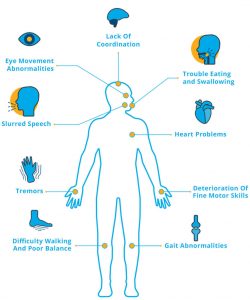
Ataxia is incoordination or clumsiness of movement. It is not due to muscle weakness. It is caused by cerebellar, vestibular, or proprioceptive sensory (large fiber/posterior column) dysfunction.
Cerebellar ataxia is produced by lesions of the cerebellum or its afferent or efferent connections. It causes irregularities in the rate, rhythm, amplitude, and force of voluntary body movements, especially at initiation and termination of motion, resulting in irregular trajectories (dysynergia), terminal tremor, and overshoot (dysmetria) in limbs. speech can become dysrhythmic (scanning dysarthria) and articulation slurred, with irregular breath control.
Vestibular ataxia has prominent vertigo (directional spinning sensations) and may cause past-pointing of limb movements, but spares speech.
Sensory ataxia has no vertigo or dizziness, also spares speech, worsens when the eyes are closed (positive Romberg sign), and is accompanied by decreased vibration and joint position sense.
Depending upon the underlying cause, Ataxias can be classified as Genetic and Acquired causes. This underlying causes help in deciding the outcome of the treatment.
Types of ataxias
The different causes of acquired ataxia can be grouped into
- Immune Mediated
- Metabolic causes
- Infectious causes
- Vascular causes
- Space occupying lesions
- Toxin / Drug induced
- Developmental anomalies
Genetic causes can be classified into
- Early onset genetic disorders (Early onset Cerebellar Ataxias)
- Spinocerebellar Ataxias (SCA)
Workup for Ataxias
Following investigations would be considered for every patient with ataxia based upon the clinical history and examination
- 1st line blood and urine studies–CBC, chemistry panel, HbA1c, fasting lipids, ESR, ANA, RPR, TSH, vitamin E, folic acid, vitamin B12, methylmalonic acid, homocysteine, urine heavy metals
- MRI brain and spinal cord, with and without contrast
- Electroencephalogram
- Evoked potentials (visual, auditory, somatosensory)
- Electronystagmogram with caloric testing
- Electromyogram with nerve conduction studies
- Chest x-ray
- 2nd line blood and urine studies – CPK, SPEP, post-prandial lactate-pyruvate-ammonia, ketones, copper, ceruloplasmin, zinc, ACE, Lyme titers, HTLV I/II, HIV, anti-thyroid antibodies, anti-gliadin antibodies (and anti-endomysial/anti-transglutaminase antibodies), anti-GAD antibodies (and antiamphiphysin antibodies)
- 3rd line blood and urine studies– very long chain fatty acids/phytanic acid, plasma or urine amino acids, urine organic acids, lysosomal hydrolase screen including hexosaminidase A, coenzyme Q10 levels, glutathione levels, PRNP gene analysis • Spinal fluid studies – cell count, glucose, lactate, protein, VDRL, gram stain, cultures as appropriate, cryptococcal antigen, 14-3-3 protein, neuron specific enolase, prion protein studies, neurotransmitter levels as appropriate, myelin basic protein, oligoclonal bands, IgG synthesis (process-specific), PCR (pathogen-specific)
- Additional imaging 1 . MR spectroscopy 2 . PEt scan/dopa-PEt scan• biopsies – conjunctival, muscle/nerve, GI tract, bone marrow, brain
- Paraneoplastic workup – appropriate imaging (ultrasound, CT, MRI), alpha fetoprotein, paraneoplastic antibodies (Yo, Hu, Ri, CV2, MaTa, Zic4, and others as available)
- Genetic workup in the ataxic patient with no family history of ataxia – in the patient over 50, occasionally positive gene tests for SCA6, SCA3, SCA1, Friedreich ataxia, and fragile X-associated tremor/ataxia syndrome (FXTAS) may be seen. Inborn errors of metabolism may occur in the patient over age 25a. Clinical whole exome sequencing can be considered